With the release of platforms like DALL-E 2 and Midjourney, diffusion generative designs have actually attained mainstream appeal, owing to their capability to produce a series of unreasonable, awesome, and frequently meme-worthy images from text triggers like “ teddy bears dealing with brand-new AI research study on the moon in the 1980s” However a group of scientists at MIT’s Abdul Latif Jameel Center for Artificial Intelligence in Health (Jameel Center) believes there might be more to diffusion generative designs than simply producing surreal images– they might speed up the advancement of brand-new drugs and minimize the possibility of unfavorable adverse effects.
A paper presenting this brand-new molecular docking design, called DiffDock, will exist at the 11th International Conference on Knowing Representations. The design’s distinct technique to computational drug style is a paradigm shift from present advanced tools that the majority of pharmaceutical business utilize, providing a significant chance for an overhaul of the conventional drug advancement pipeline.
Drugs generally operate by connecting with the proteins that comprise our bodies, or proteins of germs and infections. Molecular docking was established to acquire insight into these interactions by anticipating the atomic 3D collaborates with which a ligand (i.e., drug particle) and protein might bind together.
While molecular docking has actually resulted in the effective recognition of drugs that now deal with HIV and cancer, with each drug balancing a years of advancement time and 90 percent of drug prospects stopping working expensive scientific trials (most research studies approximate typical drug advancement expenses to be around $1 billion to over $2 billion per drug), it’s no surprise that scientists are searching for much faster, more effective methods to sort through possible drug particles.
Presently, most molecular docking tools utilized for in-silico drug style take a “tasting and scoring” technique, looking for a ligand “posture” that finest fits the protein pocket. This lengthy procedure examines a a great deal of various positions, then ratings them based upon how well the ligand binds to the protein.
In previous deep-learning services, molecular docking is dealt with as a regression issue. Simply put, “it presumes that you have a single target that you’re attempting to enhance for and there’s a single right response,” states Gabriele Corso, co-author and second-year MIT PhD trainee in electrical engineering and computer technology who is an affiliate of the MIT Computer System Sciences and Expert System Lab (CSAIL). “With generative modeling, you presume that there is a circulation of possible responses– this is crucial in the existence of unpredictability.”
” Rather of a single forecast as formerly, you now permit several positions to be forecasted, and every one with a various likelihood,” includes Hannes Stärk, co-author and first-year MIT PhD trainee in electrical engineering and computer technology who is an affiliate of the MIT Computer System Sciences and Expert System Lab (CSAIL). As an outcome, the design does not require to jeopardize in trying to reach a single conclusion, which can be a dish for failure.
To comprehend how diffusion generative designs work, it is valuable to discuss them based upon image-generating diffusion designs. Here, diffusion designs slowly include random sound to a 2D image through a series of actions, ruining the information in the image till it ends up being absolutely nothing however rough fixed. A neural network is then trained to recuperate the initial image by reversing this noising procedure. The design can then produce brand-new information by beginning with a random setup and iteratively getting rid of the sound.
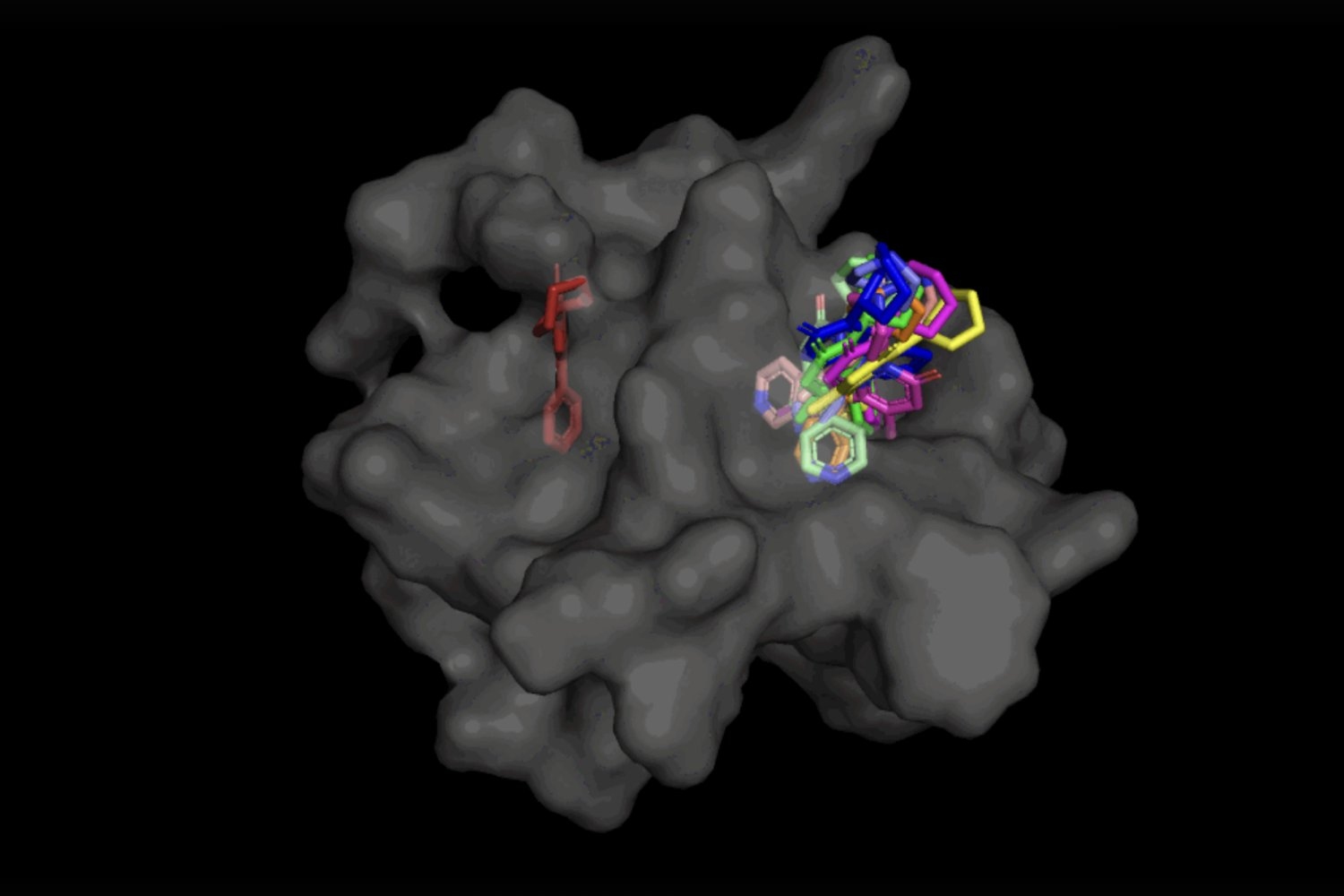
When it comes to DiffDock, after being trained on a range of ligand and protein positions, the design has the ability to effectively determine several binding websites on proteins that it has actually never ever come across prior to. Rather of creating brand-new image information, it produces brand-new 3D collaborates that assist the ligand discover possible angles that would permit it to suit the protein pocket.
This “blind docking” technique develops brand-new chances to make the most of AlphaFold 2 (2020 ), DeepMind’s popular protein folding AI design. Given that AlphaFold 1’s preliminary release in 2018, there has actually been a lot of enjoyment in the research study neighborhood over the capacity of AlphaFold’s computationally folded protein structures to assist determine brand-new drug systems of action. However advanced molecular docking tools have yet to show that their efficiency in binding ligands to computationally forecasted structures is any much better than random possibility
Not just is DiffDock substantially more precise than previous methods to conventional docking standards, thanks to its capability to factor at a greater scale and implicitly design a few of the protein versatility, DiffDock keeps high efficiency, even as other docking designs start to stop working. In the more practical situation including making use of computationally produced unbound protein structures, DiffDock positions 22 percent of its forecasts within 2 angstroms (commonly thought about to be the limit for a precise posture, 1Ã represents one over 10 billion meters), more than double other docking designs hardly hovering over 10 percent for some and dropping as low as 1.7 percent.
These enhancements develop a brand-new landscape of chances for biological research study and drug discovery. For example, numerous drugs are discovered through a procedure referred to as phenotypic screening, in which scientists observe the results of a provided drug on an illness without understanding which proteins the drug is acting on. Finding the system of action of the drug is then crucial to comprehending how the drug can be enhanced and its possible adverse effects. This procedure, referred to as “reverse screening,” can be exceptionally difficult and expensive, however a mix of protein folding methods and DiffDock might permit carrying out a big part of the procedure in silico, permitting possible “off-target” adverse effects to be determined early on prior to scientific trials occur.
” DiffDock makes drug target recognition a lot more possible. Prior to, one needed to do tiresome and expensive experiments (months to years) with each protein to specify the drug docking. And now, one can evaluate numerous proteins and do the triaging essentially in a day,” Tim Peterson, an assistant teacher at the University of Washington St. Louis School of Medication, states. Peterson utilized DiffDock to identify the system of action of an unique drug prospect dealing with aging-related illness in a current paper. “There is an extremely ‘fate enjoys paradox’ element that Eroom’s law– that drug discovery takes longer and costs more cash each year– is being resolved by its name Moore’s law– that computer systems get faster and more affordable each year– utilizing tools such as DiffDock.”
This work was performed by MIT PhD trainees Gabriele Corso, Hannes Stärk, and Bowen Jing, and their consultants, Teacher Regina Barzilay and Teacher Tommi Jaakkola, and was supported by the Artificial intelligence for Pharmaceutical Discovery and Synthesis consortium, the Jameel Center, the DTRA Discovery of Medical Countermeasures Versus New and Emerging Dangers program, the DARPA Accelerated Molecular Discovery program, the Sanofi Computational Antibody Style grant, and a Department of Energy Computational Science Graduate Fellowship.